About Idiopathic Pulmonary Fibrosis


Abbou Idiopathic Pulmonary Fibrosis (IPF)
Fibrosis leads to the gradual stiffening and thickening of lung tissue, reducing lung elasticity and making breathing difficult. In severe cases, patients may require a lung transplant.
Idiopathic Pulmonary Fibrosis (IPF) is a chronic, progressive lung disease and the most common and severe form of idiopathic interstitial pneumonia, primarily affecting men aged 50 to 70 years and older. Fibrosis causes the lung tissue to gradually stiffen and thicken, leading to reduced lung elasticity and difficulty in breathing.
The cause of IPF is unknown, which is why it is termed “idiopathic.” The fibrosis leads to the formation of persistent scarring in the lungs, giving the lungs a sponge-like appearance, which has earned it the nickname “honeycomb lung.” This damage is irreversible and progressively worsens over time, although certain medications can slow the disease’s progression. In some severe cases, patients may require a lung transplant.
The median overall survival after diagnosis is approximately 2-3 years, with a 5-year survival rate of 20%, though this varies across different countries. In Japan, the median overall survival for IPF patients is 35 months, while the 5-year survival rate in Western countries ranges from 20% to 40%. Even lower than the 5-year cancer survival rate in Taiwan (which exceeds 60%). Research indicates that after 2000, the age-standardized mortality rate for IPF was 0.5 to 12 cases per 100,000 people annually, and as of 2010, there was no increasing trend in survival rates for IPF patients, although this may have improved since then. Patients who take anti-fibrotic medications have significantly better long-term survival rates than those who do not, highlighting the importance of developing anti-fibrotic drugs.
Is there a Gender Difference in Idiopathic Pulmonary Fibrosis (IPF)? A retrospective study of 6,666 IPF patients from the UK found gender differences in baseline characteristics and prognostic factors. Males (78% of the cohort) had higher rates of smoking and comorbidities (such as ischemic heart disease and diabetes), and a lower baseline Forced Vital Capacity (FVC). Survival analysis showed that a lower Diffusing Capacity of the Lung for Carbon Monoxide (DLCO) was a worse survival predictor in males.
In the United States, between 2004 and 2017, the mortality rate decreased from 4.22 to 3.64 per 100,000 people annually, possibly due to a decline in smoking rates or changes in other environmental and genetic factors.
Symptoms of IPF

The symptoms of IPF typically develop gradually and worsen over time.
The main symptoms include:
- Persistent dry cough: A dry cough without phlegm is a typical symptom of IPF, often lasting for weeks or even months.
- Shortness of breath: Patients may experience difficulty breathing during physical activities or daily tasks.
- Fatigue: Due to reduced oxygen supply in the lungs, patients often feel unusually tired and weak.
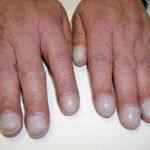
- Changes in nails and fingertips: Some patients may develop clubbing of the fingers or thickening of the nails. (picture: wikipedia)
Chronic cough, dry cough, and shortness of breath also occur in many chronic respiratory diseases such as asthma and chronic obstructive pulmonary disease (COPD), making early diagnosis challenging. According to epidemiological data from Germany, 85.9% of patients present with shortness of breath, 74.7% have chronic cough, and 40% exhibit symptoms of fatigue. The primary symptoms include progressively worsening respiratory difficulty, accompanied by restrictive ventilatory dysfunction and impaired gas exchange, leading to hypoxemia and even respiratory failure. Since the disease is confined to the lungs, its pathological and imaging features closely resemble those of usual interstitial pneumonia (UIP).
High-risk groups should undergo regular pulmonary function tests. Additionally, they can monitor their condition by observing their daily walking pace or performing the “6-Minute Walk Test.” If they notice a slower walking speed or experience shortness of breath, they should seek medical attention promptly.
What is the 6-Minute Walk Test?
The 6-Minute Walk Test involves walking back and forth over a distance of approximately 30 meters at the fastest pace possible within six minutes, without running. If you feel tired or experience leg fatigue during the test, you may slow down or take a break. If you feel unwell, the test should be stopped immediately. Throughout the six minutes, the total distance walked, as well as heart rate, blood pressure, respiratory rate, and changes in oxygen saturation, will be recorded.

Shortness of breath

Chronic cough

Fatigue
Causes and Risk Factors of IPF

The exact cause of IPF is unknown, but several risk factors may be associated with the disease:
- Age: IPF is most common in adults over the age of 50.
- Gender: Men are at a higher risk of developing IPF than women.
- Smoking history: Smoking is a significant risk factor for IPF, and the risk persists even years after quitting.
- Environmental factors: Long-term exposure to certain industrial dust or harmful substances (such as asbestos or silica dust) may increase the risk.
- Family history: A family history of IPF may increase the likelihood of developing the disease.
- Medical History: Patients with autoimmune diseases are at high risk of pulmonary fibrosis, including those with conditions such as scleroderma, rheumatoid arthritis, Sjögren’s syndrome, dermatomyositis, and polymyositis. Therefore, if symptoms of breathing difficulties arise, it is essential to maintain communication with a physician to ensure the condition does not progress further into pulmonary fibrosis.
Diagnosis of IPF
Diagnosing IPF typically requires a comprehensive assessment of multiple factors, including:
- Medical history and physical examination: Doctors will ask detailed questions about the patient’s symptoms, medical history, and family history, and perform a physical examination.
- Imaging tests: High-resolution computed tomography (HRCT) is the primary tool for diagnosing IPF, as it can reveal the characteristics of lung fibrosis.
- Pulmonary function tests: These tests assess lung function and oxygen exchange capacity.
- One-Stop Lung Health Screening: In October 2024, Taichung Veterans General Hospital launched the “Healthy Lung – Digital Dynamic Physiological Monitoring and Continuous Care Clinical Site,” integrating 7 related examinations. This provides the public with a one-stop smart medical screening station for lung diseases and comorbidities including cardiovascular, metabolic, and sarcopenia conditions while waiting for consultation (examinations that previously took an average of 10 days to complete can now be finished in less than 60 minutes) (Chinese News)
Current Treatments
Due to the limited effectiveness of existing medications and their minimal impact on improving patient survival and quality of life, many researchers are actively seeking more effective drugs and treatment methods.

Esbriet® (Pirfenidone)
- On October 15, 2014, Esbriet® (pirfenidone), a fibrosis inhibitor developed by Roche, was approved by the FDA for market release.
- Pirfenidone, with its pyridine chemical structure, is an oral fibrosis inhibitor that modulates various cytokines (e.g., INF-α, IL-1, IL-6) to promote the production of anti-inflammatory cytokine (IL-10) and inhibit the reduction of IFN-γ, thereby improving the balance between Th1 and Th2 cells. It also regulates fibrosis-related growth factors (TGF-β1, b-FGF, PDGF) to exert anti-inflammatory, anti-fibrotic, and antioxidant effects. Additionally, it inhibits fibroblast growth and collagen production. (Source: Package Insert)
- Common side effects of pirfenidone include photosensitivity reactions, rashes, loss of appetite, stomach discomfort, and nausea.
- Sandoz has announced the launch of its first generic pirfenidone in the US for treating idiopathic pulmonary fibrosis (IPF) in May 2022.

Ofev® (nintedanib)
On October 15, 2014, Ofev® (nintedanib), a multi-kinase inhibitor developed by Boehringer Ingelheim, was approved by the FDA for market release.
Ofev® is a multi-targeted kinase inhibitor that specifically inhibits the vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR). It received FDA designations such as Fast Track, Priority Review, Breakthrough Therapy, and Orphan Drug status.
The most common side effects in patients are mild to moderate diarrhea, nausea, and vomiting, which can be managed with symptomatic treatment or dosage adjustments.
- Lotus Pharmaceutical has received tentative approval from the US FDA for its Abbreviated New Drug Application (ANDA) for Nintedanib Capsules in January 2023, a generic version of Boehringer Ingelheim’s Ofev®.
- MNKD-201, developed by Mannkind, is an inhaled formulation of nintedanib. In a Phase 1 clinical trial, it was found to be safe and well-tolerated in healthy volunteers, without the typical adverse events commonly seen with oral nintedanib.

JASCAYD® (Nerandomilast)
- Nerandomilast is an oral, small-molecule chemical compound developed by Boehringer Ingelheim. It is a phosphodiesterase 4B (PDE4B) inhibitor for the treatment of Idiopathic Pulmonary Fibrosis (IPF), and was approved by the FDA and NMPA in China in October 2025.
- In September 2024, Boehringer Ingelheim announced that a global, multicenter Phase III clinical trial named FIBRONEER™-IPF had achieved its primary efficacy endpoint, showing a significant difference in the change in forced vital capacity (FVC [mL]) at Week 52 compared to the placebo group.
- The company plans to submit a New Drug Application (NDA) for nerandomilast to the U.S. Food and Drug Administration (FDA) and other global health authorities for the treatment of IPF. (Source: News)
- In 2022, the FDA granted nerandomilast Breakthrough Therapy designation for IPF.
- In February 2025, it was announced that the Phase 3 clinical trial had reached its primary clinical endpoint, with the results expected to be published in Q2 2025. (Source: News)
- In May 2025, the results of a Phase III clinical trial for Nerandomilast were announced, showing that the drug significantly slowed the decline in FVC at Week 52 and was associated with a lower mortality rate compared to the placebo group. New drug applications for the treatment of IPF and PPF have been submitted in the United States, China, and the European Union, with additional submissions planned in other regions. (Source: News)
Drugs in Development

Rentosertib (ISM001-055)
In September 2024, the results of the Phase IIa clinical trial were announced, showing positive outcomes. The 12-week study indicated that ISM001-055 demonstrated good safety and a dose-dependent response in improving lung function in IPF patients.
ISM001-055 is a chemical compound designed by Insilico Medicine through AI-based algorithms. Its mechanism of action involves targeting and inhibiting TNIK (Traf2/NCK-interacting kinase) to halt or reverse the fibrosis process, offering disease-modifying treatment for IPF patients.
>>news
AK3280
In August 2024, news revealed that the Phase II clinical trial conducted in China for AK3280 has completed patient enrollment.
Clinical Trial Results: The high-dose group of AK3280 (400 mg BID) showed a significant improvement in forced vital capacity (FVC) over 24 weeks, along with enhancements in DLco (diffusing capacity for carbon monoxide), SGRQ (St. George’s Respiratory Questionnaire), and 6MWT (6-minute walk test), with no significant adverse effects. This study is the first to confirm the safety and efficacy of AK3280 in Chinese IPF patients (Chinese News).
AK3280 is a clinical-stage drug developed by Aikobio, designed for the treatment of idiopathic pulmonary fibrosis (IPF). This drug is an optimized version of the already marketed drug pirfenidone. It can modulate multiple pathways and biomarkers closely related to the fibrotic pathological process, including the expression of fibrosis-related genes and proteins induced by transforming growth factor (TGF-β) and lysophosphatidic acid (LPA). AK3280 reduces cell proliferation, inhibits the synthesis and accumulation of extracellular matrix, and thereby exerts its therapeutic effect on IPF.
>> news

EF-011
EF-011 is an investigational drug developed by Everfront Biotech for the treatment of idiopathic pulmonary fibrosis (IPF), currently in the preclinical research phase.
Our research has identified a specific binding site for SOX2 in the promoter of type I collagen. EF-011 can reduce the binding of SOX2 to the type I collagen promoter, thereby inhibiting the expression and accumulation of type I collagen. In a bleomycin-induced mouse model of pulmonary fibrosis, treatment with EF-011 significantly improved tissue pathology and respiratory function. Functional assessments, including respiratory rate, cumulative ventilation, maximum inspiratory volume, and maximum expiratory volume, showed that the therapeutic index of EF-011 is 20 times that of the small molecule pulmonary fibrosis drug pirfenidone.
More drug development information >> EF-011
More Clinical Trial Information
- Tvardi Therapeutics: The drug TTI-101 (alone or in combination with nintedanib) for Idiopathic Pulmonary Fibrosis (IPF), failed to meet its primary objectives in the Phase 2 clinical trial data announced on October 14, due to a treatment arm discontinuation rate exceeding 50% (primarily driven by gastrointestinal adverse events) and no significant difference in FVC (Forced Vital Capacity) being observed. (Source: News)
- Pliant Therapeutics: The company terminated a Phase 2B clinical trial named Beacon-IPF based on the recommendation of the DSMB. PLN-74809 is an oral small molecule that acts as a dual selective inhibitor of αVß6 and αVß1 integrins. (Source: News)
On April 14, 2025, Bridge Biotherapeutics announced that its IPF drug candidate, BBT-877, did not meet the primary efficacy endpoint in its global Phase II clinical trial, showing no significant improvement in forced vital capacity (FVC) at Week 24. The company plans to further analyze responses in specific patient subgroups. BBT-877 is a selective inhibitor of autotaxin, a protein associated with pulmonary fibrosis. In 2019, the drug was licensed to Boehringer Ingelheim in a deal worth up to €1.1 billion, but the rights were returned a year later due to concerns about toxicity. (Source: News)
- On April 8, 2025, a study reported that Admilparant (BMS-986278), an oral lysophosphatidic acid receptor 1 (LPA1) antagonist developed by Bristol Myers Squibb, may help slow disease progression in patients with idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). Compared to placebo, IPF patients treated with Admilparant had a lower risk of disease progression over 26 weeks, with the 60 mg dose showing greater efficacy than the 30 mg dose. (Source: News)
- On September 3, 2025, new clinical data for the inhaled therapy Tyvaso® (treprostinil) showed significant benefits for patients with idiopathic pulmonary fibrosis (IPF). After 52 weeks of treatment, patients demonstrated greater improvements in lung function, measured by forced vital capacity (FVC), compared with placebo. Improvements were also observed in quality of life and other pulmonary function measures. In terms of safety, Tyvaso was well tolerated overall, with no new safety concerns reported. (Source: News)
Care and Precautions for IPF Patients

Oxygen Supplementation
Pulmonary fibrosis patients often experience difficulty breathing. When blood oxygen levels are low, oxygen therapy can help patients maintain mobility and comfort.

Pulmonary Rehabilitation Program
Pulmonary rehabilitation programs typically include exercise training, nutritional guidance, and psychological support, which can help improve patients’ quality of life.
The University of Pittsburgh conducted a 12-week clinical trial involving 63 patients with pulmonary fibrosis, using a modified yoga program specifically designed for this patient group. The program included seated poses, breathing exercises, and meditation. Results showed improvements in health-related quality of life, with particularly notable relief in coughing symptoms. The study indicated that posture correction and breath control techniques can enhance the efficiency of diaphragmatic and abdominal breathing, helping to manage shortness of breath and reduce cough reflex sensitivity.

Maintain a Healthy Diet
Due to the difficulty in breathing experienced by patients with pulmonary fibrosis, they often suffer from symptoms such as wheezing and coughing. Over time, this makes it difficult for them to eat, which can lead to malnutrition and even conditions like sarcopenia. Doctors recommend that patients consume high-protein and high-calorie foods to ensure they have enough energy and heat. They should also avoid foods that may trigger allergies or eating too much, as it can cause the diaphragm to be compressed and lead to shortness of breath.

Maintaining Quality of Life
The prognosis of IPF varies among individuals, but the disease usually worsens over 3 to 5 years. With appropriate treatment, many patients can still maintain a certain quality of life.

Regular Medical Visits and Follow-Up
Patients should closely collaborate with their healthcare team to develop a personalized treatment plan and undergo regular follow-ups to monitor disease progression.

Managing Cardiovascular Disease (CVD)
A systematic review and meta-analysis of 28 studies showed that patients with idiopathic pulmonary fibrosis (IPF) have a significantly increased risk of cardiovascular disease (CVD). The risk of developing ischemic heart disease, thromboembolic disease, pulmonary hypertension, and other forms of heart disease was elevated by approximately 1.1- to 2.5-fold. The study concluded that systematic cardiovascular risk screening and management should be strengthened in IPF patients to improve treatment tolerability and health outcomes.